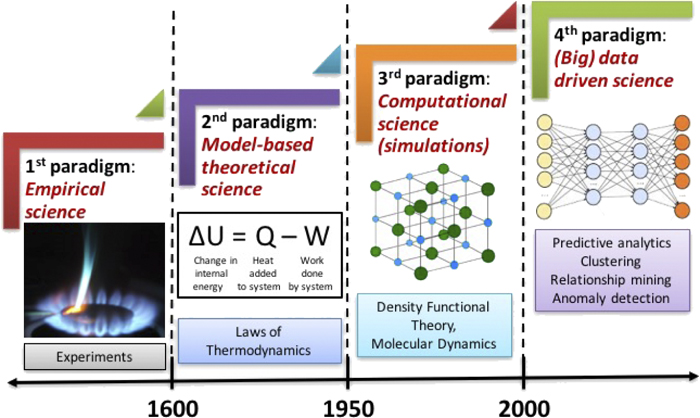
Lecture 8#
Electron density \(\mathbf{\rho(r)}\)
For a multi-electron wave function \(\Psi(x_1, x_2, \ldots, x_N) \rightarrow \underset{\underset{x,y,z}{\text{Spatial}}}{3N} + \underset{\text{Spin}}{N}\) variables \(\rightarrow\) It is quire complex!
So, we use density to describe it:
\(\rho(r\rightarrow\infty)=0\)
\(\int\rho(r)dr=\underset{\text{num of electrons}}{N}\)
Functional: function of functions
Input |
Output |
Example |
|
|---|---|---|---|
Function |
Number |
Number |
\(f(x)=\sin(x)\) |
Functional |
function |
Number |
\(F[\rho(r)]\), \(\int_{-\pi}^{\pi}\sin(x)dx\) |
Operator |
function |
function |
\(H\Psi= \hat{H}\Psi\) |
1. The Hohenberg-Kohn Theorems#
Theorem I: For any system of interacting particles in an external potential ( V_{\mathrm{ex}}(\mathbf{r}) ), the potential ( V_{\mathrm{ex}}(\mathbf{r}) ) is determined uniquely, except for a constant, by the ground-state particle density ( \rho_0(\mathbf{r}) ).
\(E_{\text{total}}\) is a function of \(\Psi\), we indicate this property using square brackets:
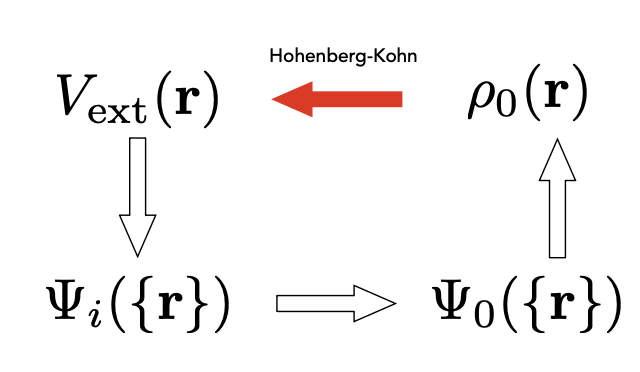
Theorem II: A universal functional for the total energy \(E[\rho]\) in terms of the density \(\rho(r)\) can be defined, valid for an external potential \(V_\text{ex}(r)\). For any particular \(V_\text{ex}(r)\), the exact ground state energy of the system is the global minimum value of this functional, and the density ρ® that minimizes the functional is the exact ground state density \(\rho_0(r)\).
2. The Kohn-Sham Auxiliary System#
Previous section describes how to represent multi-electron system into density form, using functional, but what functional looks like? How to solve it?
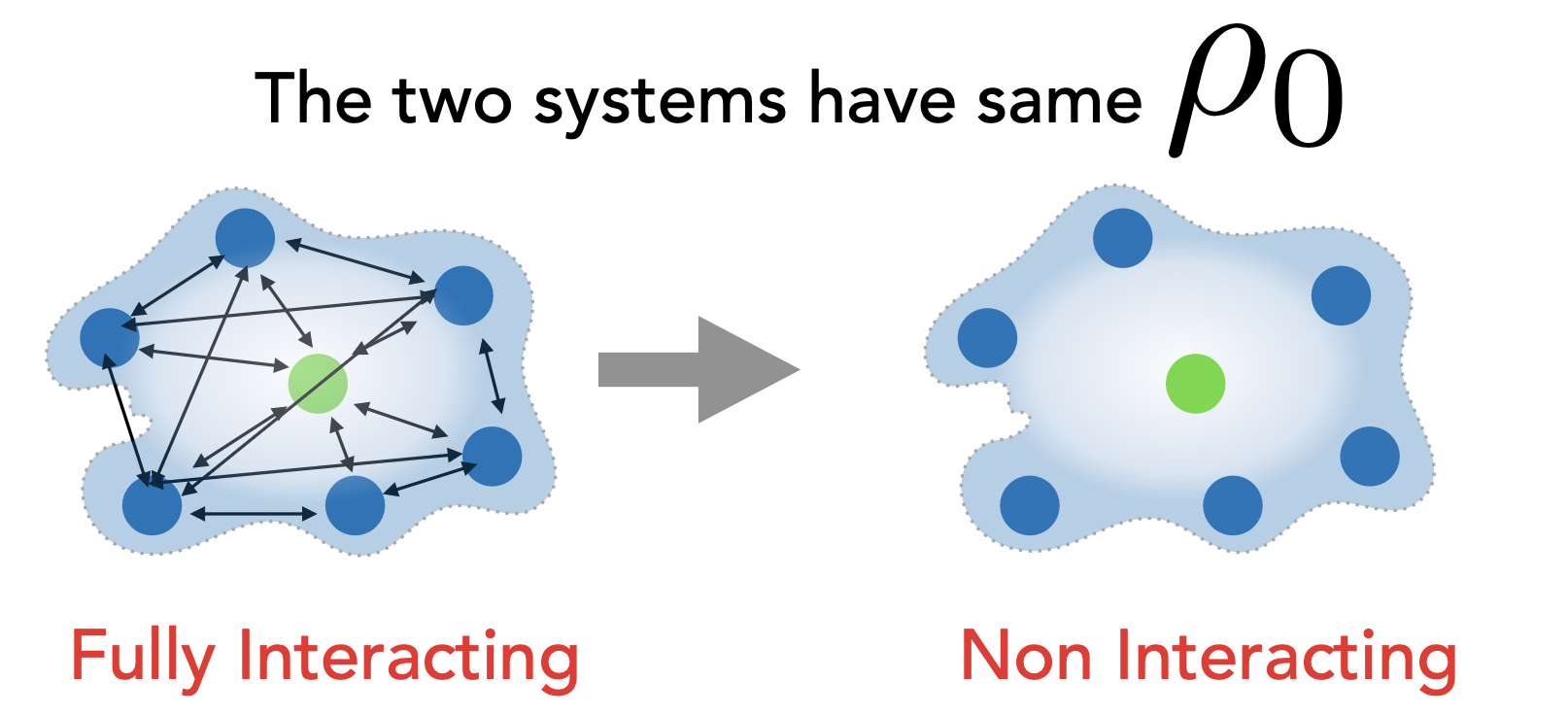
[!NOTE]
Same \(\rho_0(r)\), but not approximation compared with Hartree-Fock method.
The actual density functional theory calculations are performed on the auxiliary single-electron independent-particle system defined by the mono-electronic auxiliary hamiltonian \(\hat{H}_\text{aux}\), also known as the Kohn-Sham operator \(\hat{F}_\text{KS}\) in \(a.u.:^2\)
[ \left{ \begin{aligned} &\text{eigenvalue}\rightarrow \theta_i® \quad\text{: K-S orbitals(one electron orbital)} \ &\text{eigenvalue}\rightarrow \epsilon_i \quad\text{:eigenvalue for orbital i} \end{aligned} \right. ]