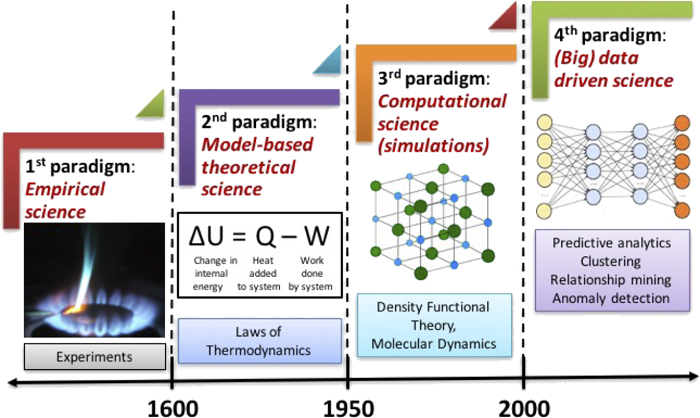
1. Introduction#
1. Characteristic length and times scales for molecular dynamics#
Types |
Time scale |
Time scale (s) |
|---|---|---|
Bond vibration |
1 fs; Femtosecond |
\(10^{-15}\) s |
Collective vibration |
1 ps; Picosecond |
\(10^{-12}\) s |
Conformational transition |
ps or longer |
\(>10^{-12}\) |
Enzyme catalysis |
\(\mu\)s to ms |
\(10^{-6}\sim 10^{-3}\) s |
Ligand binding |
\(\mu\)s to ms |
\(10^{-6}\sim 10^{-3}\) |
Protein folding |
ms; millisecond to s |
\(10^{-3}\sim 10^0\) s |
For an atomistic simulation, it typically use an iteration time step of 1 fs (to capture bond vibrations)
And the accessible time scale for us with atomistic simulation MD is \(100\text{ns} = 10^5 \text{ ps}=10^8 \text{ fs}=10^{-7} \text{s }\)
2. Empirical potential energy#
2.1. Lennard Jones potential#
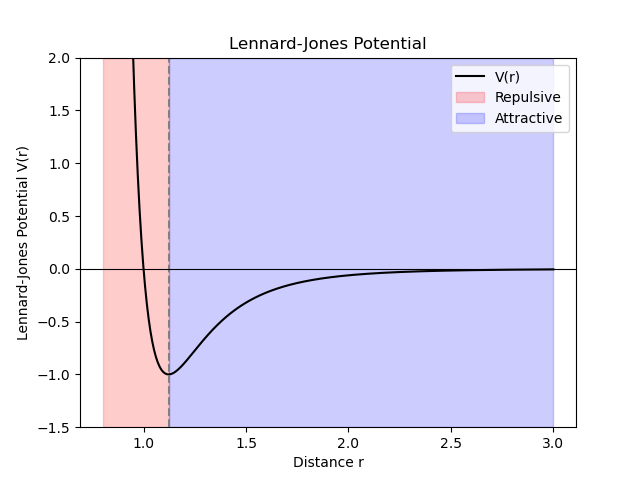
\(\sigma\) is the unit of length scale
\(\epsilon\) is the unit of energy scale
2.2. Morse potential#
 $$
\begin{aligned}
V(r) &= D[1-e^{(-\alpha \cdot (r - r_0))}]^2 - D \\
&= D[\exp(-2\alpha \cdot (r - r_0)) - 2\exp(-\alpha \cdot (r - r_0))]
\end{aligned}
$$
$$
\begin{aligned}
V(r) &= D[1-e^{(-\alpha \cdot (r - r_0))}]^2 - D \\
&= D[\exp(-2\alpha \cdot (r - r_0)) - 2\exp(-\alpha \cdot (r - r_0))]
\end{aligned}
$$
D is the unit energy scale, also often refers to bond dissociation energy
\(\alpha\) is the elastic properties, or force constant (the spring constant) of the bond
\(r_0\) is the equilibrium distance.
2.3. Buckingham potential#
\(\frac{D}{r^8}\): sometimes a \(2^{nd}\) order term is added to satisfy van der Waals perturbation theory (\(r^{-8}\))
Fundamental issues of pair potentials
[!NOTE]
Pair potentials can not “count bonds”, and do not care about the organization of atoms (angles, etc.)
In pair potential models the cohesive energy on an atom is largely determined by how many bonding partners are around the atom