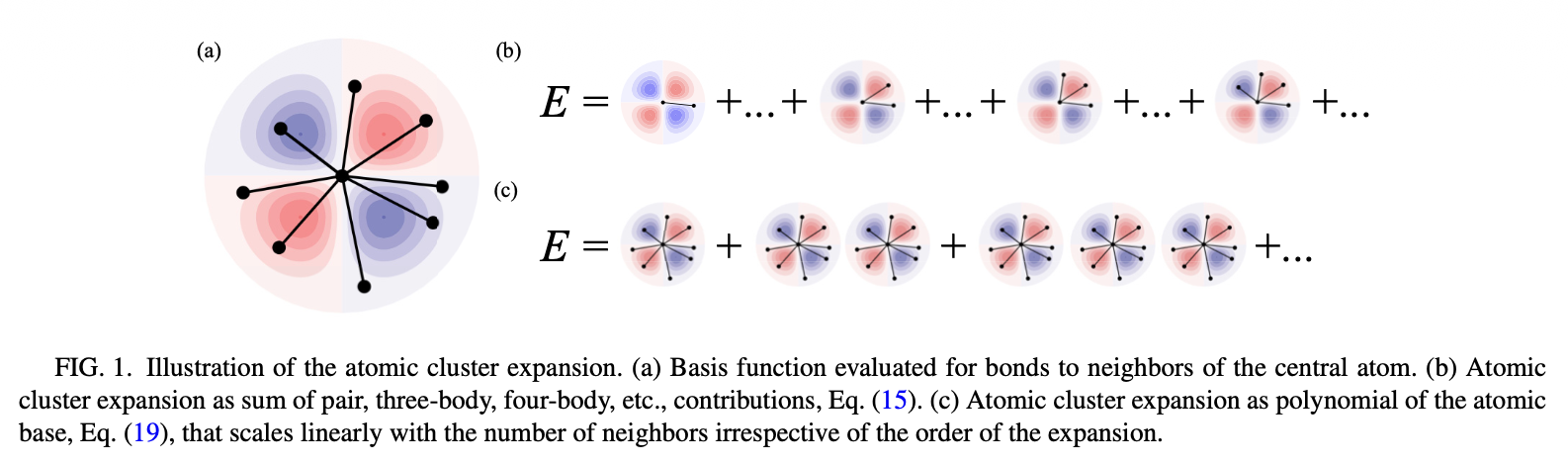
Atomic Cluster Expansion (ACE)#
Background
The energy of a system can be assumed that the total energy E of the system as a sum of atomic contributions \(E_i\)
\(r_i\) is the position of atom \(i\) and the potential \(V^{(2)}, V^{(3)},...\) are symmetric, uniquely defined, and zero if two or more indices take identical values. Commonly, \(V_0\) is a constant offset that can be set to zero and \(V^{(1)}\) is the chemical potential
So, the above equation can be written into atomic contributions for each separated energy:
As you can see here, such atomic expansion requires higher order Tylor expansion, the convergence is slow. E,g. Bulk metal potential \(V^{(K)}\) up to \(K>15\) is required. Even with cutoff that just taking account of nearby atoms within cutoff \(r_c\), the evaluation of the leading termof order \(K + 1\) in the sum Eq. (3) scales as \(N_c^K\), where \(N_c\) corresponds to a typical number of neighbors within the cutoff sphere. e.g. for an accurate potentials one requires cutoffs that in a closed packed materials imply \(N_c ≈ 10^2 ... 10^3\). It’s challenging to sum expansions within acceptable time.
1. ATOMIC CLUSTER EXPANSION#
We first define the inner product in Hilbert space:
introduced:
[!NOTE]
Proof of completeness: Set of basis functions \(\phi_v\) is capable of describing any possible local atomic environment.
If the basis is complete: \(f(r)=\sum_v c_v\phi_v(r)\), where \(c_v\) is the coefficient for the basis function
To find a specific coefficient, we project \(f(r)\) into the basis, which is the inner product, so, \(c_v=\int \phi_v^\star(r^\prime)f(r^\prime)dr^\prime\)
The we substitute the \(c_v\) obtained by projection to the original formula: \(f(r)=\sum_v\Big[\int \phi_v^\star(r^\prime)f(r^\prime)dr^\prime\Big]\phi_v(r)=\sum_v\int[\phi_v^\star(r^\prime)\phi_v(r)]f(r^\prime)dr^\prime\)
And \(\phi_v^\star(r^\prime)\phi_v(r)=\delta(r^\prime-r)\)
Define the cluster expansion
Here we define the basis functions for the expansion of the atomic energy from the product of single-bond basis functions. By choosing \(\phi_0= 1\), a hierarchical expansion is obtained.
A cluster \(\alpha\) with \(K\) elements contains \(K\) bonds \(\alpha = (j_{1i},j_{2i},. . . , j_{Ki})\), where the order of entries in \(\alpha\) does not matter, and the vector \(v = (v_1,v_2,. ..,v_K )\) contains the list of single-bond basis functions in the cluster. Only single-bond basis functions with v > 0 are considered in ν. The cluster basis function is given by: